
Пациент — мужчина, 43 лет
нечеткость речи, сонливость, одышка при длительной ходьбе, общая слабость, быстрая утомляемость, боли и судороги в мышцах голеней после длительной ходьбы.
Семейный анамнез: мать страдает от мигрени, катаракта с 62 лет; у отца с 59 лет ИБС.
Анамнез: раннее моторное и психо-речевое развитие по возрасту. В школе и молодости занимался спортом, но особых достижений не имел. Отмечает, что никогда не умел свистеть.
20 лет: стал хуже бегать, бросил футбол;
24 года: боли в спине, обследован, установлен диагноз «остеохондроз»;
26 лет: нарушения походки, трудно долго ходить и подниматься по лестнице;
30-35 лет: прогрессируют нарушения сна, усилился ночной храп (со слов родственников), выраженная сонливость днем, частые головные боли.
Другие хронические заболевания у себя пациент отрицает, у врачей не наблюдается.
Результаты обследований:
УЗИ ОБП: без патологии.
ЭКГ: синусовая тахикардия 96/мин, неполная блокада правой ножки пучка Гисса.
ФЖЕЛ: ↓16% в положении лежа на спине от значений в положении сидя.
Полисомнография: ночная гиповентиляция, эпизоды гипоксемии, индекс апноэ/гипопноэ 8/ч (признаки обструктивного апноэ сна).
Биохимический анализ крови: КФК 815 Ед/л (↑), АЛТ 125 Ед/л и АСТ 93 Ед/л (↑); билирубин/ГГТ норма.
Стимуляционная ЭНМГ: без особенностей.
Игольчатая ЭНМГ: миотонические разряды в бицепсе и параспинальных мышцах; ПДЕ: средняя длительность снижена, амплитуда умеренно увеличена, больше данных за первично-мышечные изменения.
Объективно: умеренная слабость мимических мышц и языка (до 3-4 б); проксимальные мышцы рук и ног до 3 б, дистальные сохранны. Умеренная атрофия дельтовидных мышц, плечевого пояса, паравертебральных, ягодичных и бедренных мышц. «Крыловидные» лопатки, поясничный гиперлордоз, выступающий живот. Походка «переваливающаяся», ходьба на носках/пятках сохранена. Сухожильные рефлексы: с рук живые, с ног снижены, D=S.
____________________________________________________________________________________
* Случай смоделирован. Любое совпадение с реальной клинической практикой — случайность
Какой диагноз лучше всего объясняет совокупность данных?
Диагноз неверный
Диагноз неверный
Диагноз неверный
Диагноз неверный
Диагноз неверный
Диагноз верный
Правильный ответ
Болезнь Помпе с поздним началом (БППН)
БППН – это аутосомно-рецессивная лизосомная болезнь накопления, которая возникает в результате недостаточной активности фермента кислой 1,4-глюкозидазы/ кислой мальтазы.
Дефицит фермента приводит к накоплению гликогена в лизосомах клеток скелетных и дыхательных мышц, миокарда, гладких мышц сосудов и внутренних органов.
Прогрессирующее депонирование гликогена вызывает постепенную деструкцию мышечных волокон и дисфункцию вовлеченных в патологический процесс органов и тканей.
Причина: биаллельные патогенные варианты в гене GAA.
Дебют: младенчество, детство или взрослый возраст (в зависимости от фенотипа).
Фенотипы:
Инфантильная форма:
Клиническая картина:
В основе — признаки проксимальной мышечной слабости, особенно в мышцах тазового пояса
Характерные лабораторные изменения:
Инструментальные исследования:
ЭНМГ: признаки первично-мышечного поражения (↓ амплитуды и длительности ПДЕ).
Также могут выявляться миотонические разряды, особенно, в параспинальных мышцах.
Дифференциальная диагностика:
Первичный скрининг и ДНК-диагностика:
- определение активности кислой 1,4-глюкозидазы в «сухих пятнах» крови,
- исследование гена GAA
Прогноз:
Основные причины летальных исходов связаны с респираторными осложнениями.
Лечение:
Патогенетическое лечение: ферментозаместительная терапия рекомбинантной кислой α-1,4-глюкозидазой.
Своевременность диагностики БП — актуальная проблема, учитывая возможности патогенетического лечения.
Дефицит фермента приводит к накоплению гликогена в лизосомах клеток скелетных и дыхательных мышц, миокарда, гладких мышц сосудов и внутренних органов.
Прогрессирующее депонирование гликогена вызывает постепенную деструкцию мышечных волокон и дисфункцию вовлеченных в патологический процесс органов и тканей.
Причина: биаллельные патогенные варианты в гене GAA.
Дебют: младенчество, детство или взрослый возраст (в зависимости от фенотипа).
Фенотипы:
Инфантильная форма:
- дебют в первые месяцы жизни (до года);
- проявляется тяжелой гипотонией, гипертрофической кардиомиопатией с кардиомегалией, задержкой моторного развития, расстройствами глотания и прогрессирующей дыхательной недостаточностью.
- дебют после 1-го года жизни;
- более медленное течение за счет частично сохранной активности фермента, отсутствие кардиомиопатии в раннем возрасте и преимущественное поражение скелетных мышц; со временем также неуклонно прогрессирует, с постепенно нарастающей слабостью скелетных мышц и дыхательной недостаточностью.
Клиническая картина:
В основе — признаки проксимальной мышечной слабости, особенно в мышцах тазового пояса
- походка «вперевалку»(по типу «утиной»);
- затруднения при ходьбе, подъеме по лестнице;
- приемы Говерса;
- общая слабость и утомляемость;
- формирование поясничного гиперлордоза, что нередко сопровождается хроническими болями в спине («остеохондроз»);
- эпизоды болезненных мышечных судорог;
- дыхательные нарушения (одышка при привычной физической активности, снижение толерантности к физическим нагрузкам, ортопноэ, ночная гиповентиляция, что сопровождается утренними головными болями и дневной сонливостью)
- ↓ ЖЕЛ, дыхательная недостаточность рестриктивного типа
- частые респираторные инфекции
Характерные лабораторные изменения:
- умеренное ↑ КФК
- ↑ АЛТ, АСТ, ЛДГ
- выраженное ↑ Glc₄ (тетрасахарид глюкозы) в моче.
Инструментальные исследования:
ЭНМГ: признаки первично-мышечного поражения (↓ амплитуды и длительности ПДЕ).
Также могут выявляться миотонические разряды, особенно, в параспинальных мышцах.
Дифференциальная диагностика:
- мышечные дистрофии (другие поясно-конечностные мышечные дистрофии, миодистрофия Дюшенна/ Беккера, миотоническая дистрофия 2 типа, лице-плече-лопаточная мышечная дистрофия Ландузи-Дежерина);
- болезни мотонейрона (СМА 5q, бульбоспинальная мышечная атрофия Кеннеди, БАС);
- метаболические миопатии (гликогенозы IIIа, IV V и VII типов);
- митохондриальные миопатии;
- воспалительные миопатии;
- болезни нервно-мышечной передачи (миастения гравис, врожденные миастенические синдромы, синдром Ламберта-Итона);
- асимптомное повышение КФК.
Первичный скрининг и ДНК-диагностика:
- определение активности кислой 1,4-глюкозидазы в «сухих пятнах» крови,
- исследование гена GAA
Прогноз:
Основные причины летальных исходов связаны с респираторными осложнениями.
Лечение:
Патогенетическое лечение: ферментозаместительная терапия рекомбинантной кислой α-1,4-глюкозидазой.
Своевременность диагностики БП — актуальная проблема, учитывая возможности патогенетического лечения.
Вернуться на страницу Клинические задачи
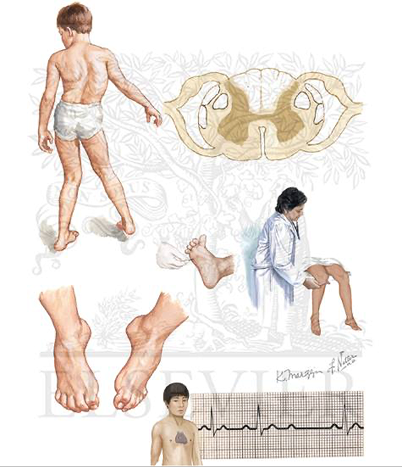
Клиническая картина к кейсу №72