
Пациент — женщина, 33 года
Жалобы: шаткость, неустойчивость при ходьбе, периодические головокружения, онемения пальцев стоп.
Анамнез: раннее развитие по возрасту;
5 лет: расходящееся косоглазие;
16-20 лет: частые мигрени;
23 года: нейросенсорная тугоухость 2 ст.;
27 лет: госпитализирована по поводу выраженной головной боли и рвоты, нарушения сознания;
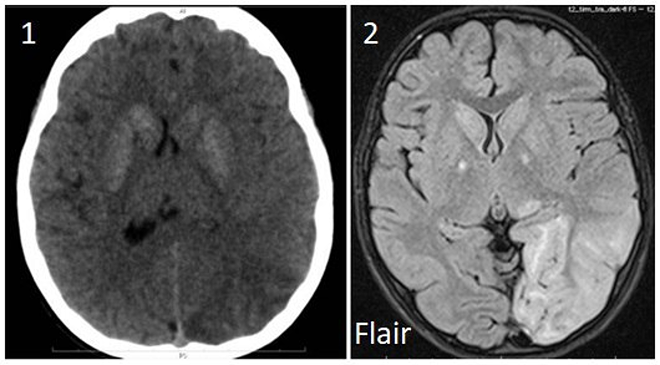
по данным КТ ГМ предположен герпетический энцефалит (посмотреть фото);
проведено лечение, симптомы купированы, но после выписки отмечали некоторую заторможенность реакций;
31 год: сахарный диабет 1 типа, отмечается высокая вариабельность гликемии до 35% в сутки;
33 года: жалобы на неустойчивость при ходьбе и головокружения;
34 года: повторная госпитализация с аналогичными жалобами
Проведено исследование спинномозговой жидкости на простой герпес, цитомегаловирус, туберкулез - отрицательно.
Антинуклеарные антитела (АНА) отрицательные.
МРТ ГМ: см. рис.2
ЭМНГ: признаки поражения периферических нервов нижних конечностей по демиелинизирующему типу с обеих сторон
Игольчатая ЭМГ: миогенный паттерн поражения
КФК: 195 Ед/л
ЭКГ: синусовая аритмия
Офтальмолог: расходящееся альтернирующее косоглазие обоих глаз, миопия слабой степени, миопический астигматизм.
Осмотр: рост 164 см, вес 54 кг. В неврологическом статусе незначительное ограничение отведения глазных яблок во все стороны, установочный нистагм, рефлексы с рук живые, с ног высокие, с расширением зон вызывания. ПНП с легкой интенцией и мимопопаданием.
____________________________________________________________________________________
* Случай смоделирован. Любое совпадение с реальной клинической практикой – случайность
О чем можно подумать?
Диагноз неверный
Диагноз верный
Диагноз неверный
Диагноз неверный
Диагноз неверный
Диагноз неверный
Правильный диагноз
MELAS
MELAS (mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes, митохондриальная энцефаломиопатия, лактат-ацидоз, инсультоподобные эпизоды) –мультисистемное заболевание, обусловленное изменениями в митохондриальной ДНК (мтДНК), с преимущественным поражением нервной системы и мышц
Митохондриальный тип наследования: по «материнской» линии (отцовские митохондрии находятся только в хвостовой части сперматозоидов, которая не проникает в яйцеклетку)
Причина: патогенные варианты генов тРНК митохондрий
Дебют: от детского до 40 лет (редко 50-60 лет)
Клиническая картина
Дебют в детском возрасте:
- период нормального психомоторного развития
- задержка роста (иногда)
- генерализованные тонико-клонические судороги
- мигренозные головные боли
- анорексия
- рвота
- непереносимость физических нагрузок
- проксимальная мышечная слабость
- лактат-ацидоз
Дебют во взрослом возрасте:
- инсультоподобные состояния
- нейросенсорная тугоухость
- когнитивные нарушения
- полинейропатия
- офтальмоплегия
- сахарный диабет
- нарушения ритма сердца
- лактат-ацидоз
Инсультоподобные состояния:
- сильная головная боль
- рвота
- расстройства сознания
- очаговая симптоматика
Возможные нарушения психических функций:
Характерно постепенное нарастание очаговых симптомов. Как правило, преходящий характер нарушений, однако повторные эпизоды могут оставлять стойкие очаговые нарушения или изменения на МРТ
Нейровизуализация (частые признаки):
КТ: кальцификаты базальных ганглиев
МРТ: очаги усиления сигнала в режимах Т2/ FLAIR (чаще в задних отделах мозга, не совпадающих с зонами васкуляризации крупных сосудов)
При использовании метода DWI (диффузно-взвешенные изображения) коэффициент диффузии повышен, в отличие от снижения при ишемическом инсульте
Дифференциальная диагностика:
Биоматериал для молекулярно-генетической диагностики:
Первый этап диагностики: исследование частых патогенных вариантов (нуклеотидные замены m.3243A>G и m.3271T>C регистрируются в большинстве случаев)
Митохондриальный тип наследования: по «материнской» линии (отцовские митохондрии находятся только в хвостовой части сперматозоидов, которая не проникает в яйцеклетку)
Причина: патогенные варианты генов тРНК митохондрий
Дебют: от детского до 40 лет (редко 50-60 лет)
Клиническая картина
Дебют в детском возрасте:
- период нормального психомоторного развития
- задержка роста (иногда)
- генерализованные тонико-клонические судороги
- мигренозные головные боли
- анорексия
- рвота
- непереносимость физических нагрузок
- проксимальная мышечная слабость
- лактат-ацидоз
Дебют во взрослом возрасте:
- инсультоподобные состояния
- нейросенсорная тугоухость
- когнитивные нарушения
- полинейропатия
- офтальмоплегия
- сахарный диабет
- нарушения ритма сердца
- лактат-ацидоз
Инсультоподобные состояния:
- сильная головная боль
- рвота
- расстройства сознания
- очаговая симптоматика
Возможные нарушения психических функций:
- деменция
- депрессия
- психоз
- тревожность
- личностные изменения
Характерно постепенное нарастание очаговых симптомов. Как правило, преходящий характер нарушений, однако повторные эпизоды могут оставлять стойкие очаговые нарушения или изменения на МРТ
Нейровизуализация (частые признаки):
КТ: кальцификаты базальных ганглиев
МРТ: очаги усиления сигнала в режимах Т2/ FLAIR (чаще в задних отделах мозга, не совпадающих с зонами васкуляризации крупных сосудов)
При использовании метода DWI (диффузно-взвешенные изображения) коэффициент диффузии повышен, в отличие от снижения при ишемическом инсульте
Дифференциальная диагностика:
- ОНМК различной этиологии
- осложненные мигрени
- детский альтернирующий паралич
- нейроинфекция
- болезнь Фабри
- гомоцистинурия
- болезнь моя-моя
- другие митохондриальные заболевания (синдром Кирнса-Сейра, синдром MERRF, синдром Ли, и другие)
Биоматериал для молекулярно-генетической диагностики:
- осадок мочи + кровь
- биоптат мышечной ткани (в случае типичной картины и отсутствии патогенных вариантов в крови и моче)
Первый этап диагностики: исследование частых патогенных вариантов (нуклеотидные замены m.3243A>G и m.3271T>C регистрируются в большинстве случаев)
Вернуться на страницу Клинические задачи
КТ ГМ предположен герпетический энцефалит